Home | Introduction | Metadata | Application | Further reading | Landscape of resources |
Metadata
- Clinical study design and FAIR implementation
- Clinical study data process through the FAIR lens
- Planning and preparation of the study
- Approvals, permissions and agreements
- Study conduct and data collection
- Data curation / harmonisation processes
- Analysis of data
- Reporting
- Data follow-up activities
- Common Data Models for clinical data
- CDISC
- OHDSI
- FHIR®
- Shared model of clinical study design and outcome
- The BRIDG initiative
- Semantic integration with Semantic Web standards
Clinical study design and FAIR implementation
Good scientific practice for life science research and development requires careful planning and design for the collection of data from any source, such as laboratory experiments, clinical studies, databases and scientific literature. A Data Management Plan (DMP) is an essential element in the process to manage the FAIR data life cycle. It will document the plan for the dataset and associated metadata in specific terms of what, how, who and when. This process will support making the data FAIR by design and is highly relevant to clinical study design. More details on the DMP method can be found in the Pistoia Alliance FAIR Toolkit: Data Management Plans
Clinical studies are traditionally described in human readable form as protocol documents, which represent one of the main types of clinical trial descriptors. Typically, a protocol for a clinical trial contains a study description, usually available as free text, a specification of the nature of the interventions and how the trial will be conducted, as well as, for example, safety concerns and other ethical considerations. However, requirements from the regulators, funding agencies and publishers now mandate moving away from textual narrative to much more syntactically and semantically structured reporting. From a general information management standpoint, but also more specifically from a FAIR implementation viewpoint, “free text” is problematic as, without elaborate methods such as text mining and manual curation to structure and meaningfully annotate a study, key information lacks machine actionability. Therefore, submitting a clinical trial to the regulatory authorities or uploading a clinical study to one of the public repositories, e.g clinicaltrials.gov, requires structuring of the data through the mandatory use of a designated format specification. For instance, in the context of regulatory submission, the CDISC SDTM clinical standard is mandated.
However, adhering to these specifications is not enough to achieve a FAIR maturity level that enables full machine actionability (see section 3 of this guide) and computable knowledge still remains out of reach for software agents even when the text description has a standardised structure. To further demonstrate the problem, we considered the following trial as our test bed: “Effect of Propolis or Metformin Administration on Glycemic Control in Patients With Type 2 Diabetes Mellitus” available from http://clinicaltrials.gov (link).
 Figure 2: This study description is taken from https://clinicaltrials.gov/ct2/show/study/NCT03416127
Figure 2: This study description is taken from https://clinicaltrials.gov/ct2/show/study/NCT03416127
The initial examination unsurprisingly reveals that the “Brief summary” of the trial is human readable and, more interestingly that the same information can also be accessed programmatically via an API providing a json file to an HTTPS request. Upon closer inspection however, the web pages for human consumption reveals that only basic metadata markup by relying on the OpenGraph protocol is provided. Search engine optimisation (SEO) using schema.org markup is absent and may somewhat limit discoverability and findability.
We then focused on the data submission process: In order to submit the trial to http://clinicaltrials.gov, the (meta)data has to be submitted in a structured way, a set of key/value pairs, which can be presented as a table containing the study protocol and summary results, in addition to the free text (see below).
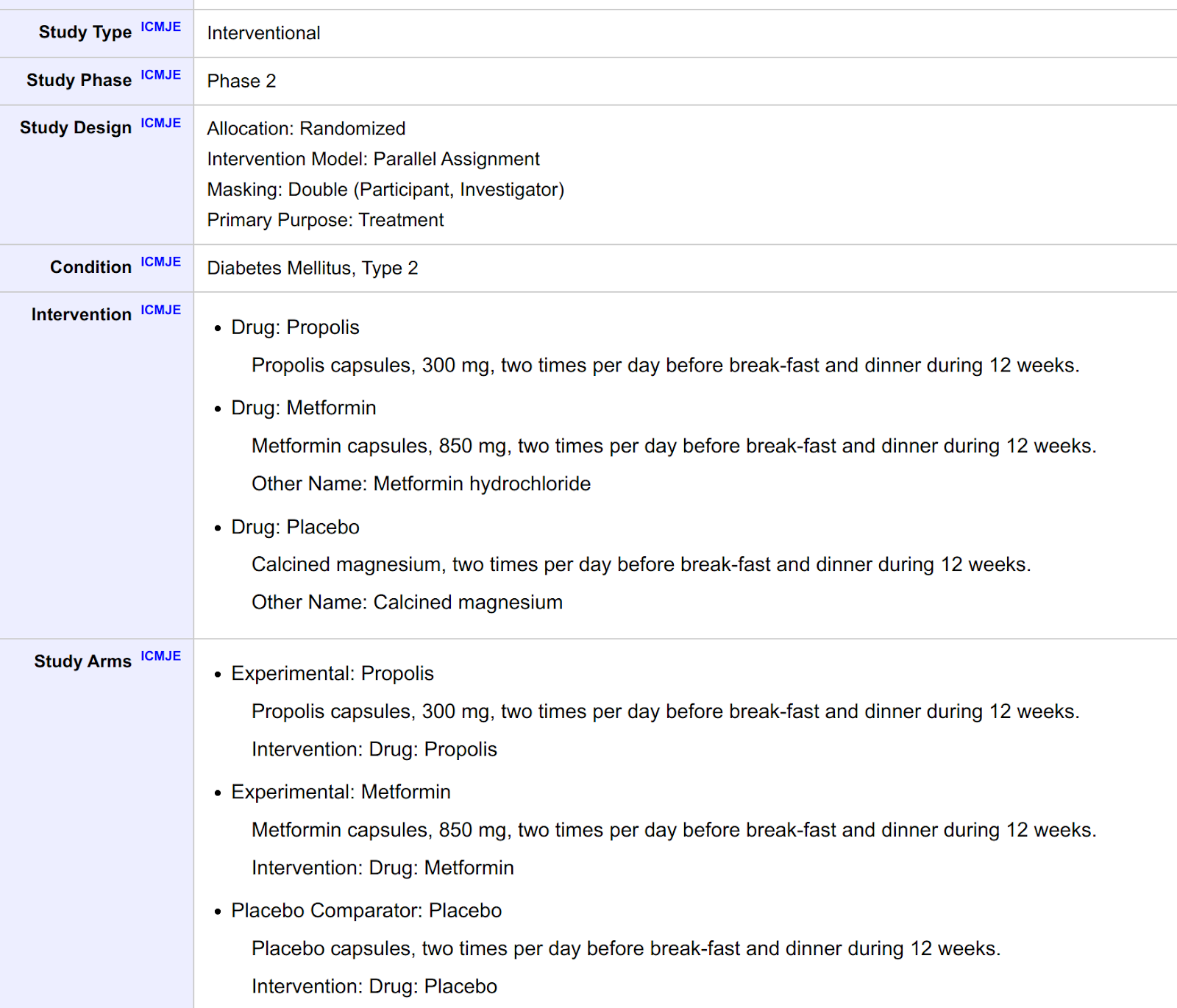 Figure 3: Structured descriptive Information (i.e. structured metadata) for https://clinicaltrials.gov/ct2/show/study/NCT03416127. Structured summary results are available as well in a separate tab.
Figure 3: Structured descriptive Information (i.e. structured metadata) for https://clinicaltrials.gov/ct2/show/study/NCT03416127. Structured summary results are available as well in a separate tab.
Whilst providing structured text, this is still insufficient to qualify as FAIR. Mature implementation of the FAIR guiding principles goes much further than this: for instance, each key should be associated with an entity from a semantic model via an uniform resource identifier (URI) that is a GUPIR and the associated value should also be marked up for indexing by search engines.
With these observations in mind and with the knowledge that FAIRifying data and metadata retrospectively can be very costly and time consuming (see the Roche use case in the Pistoia Alliance FAIR Toolkit - FAIRification of clinical trial data), the following sections will expand on the notion of prospective FAIRification of data and metadata. This approach, to implement the FAIR guiding principles from the start to facilitate secondary reuse and by design, saves costs, ensures efficiency and supports longevity of data and metadata. We explore the key aspects of prospective FAIRification of data and metadata from clinical studies in the following chapter “Clinical study data process through the FAIR lens”.
Clinical study data process through the FAIR lens
Data collection in a clinical study is usually highly regulated and thus a sequential process. It starts with planning and preparation, obtaining legal approvals, conducting the study and primary and secondary data analysis (see figure 4). In this section we want to briefly discuss each step and especially highlight which aspects of FAIR are important to consider and how FAIR data can be beneficial in later steps of the process.
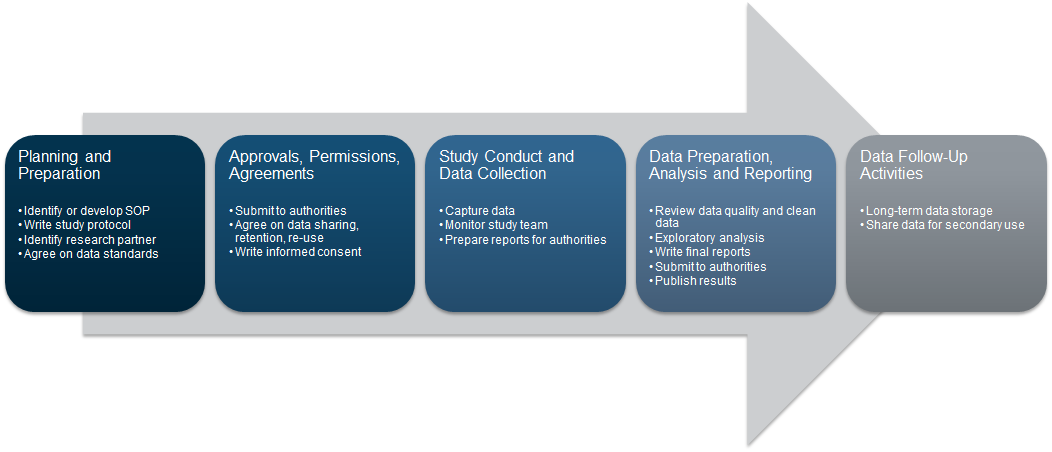 Figure 4: Adapted from nidcd.nih.gov
Figure 4: Adapted from nidcd.nih.gov
Planning and preparation of the study
Prospective FAIRification means moving a number of processes upstream, before the start of the data collection process. How data is collected should be defined in the FAIR Data Management Plan with the FAIR principles implemented to enable future re-use of the data and metadata. For example, domain specific standards and vocabularies should be clearly identified and incorporated as required for interoperability as defined by the FAIR principles.
FAIR implementation: Interoperability, use of syntax standards and vocabularies
Approvals, permissions and agreements
Before a clinical study can go ahead, a range of approvals (e.g. regulatory, ethical) need to be obtained and potential restrictions (e.g. in terms of access and data use) need to be clarified so an informed consent form can be created. Data management plans need to define storage, retention periods, condition of access and consider aspects of re-use and data-sharing. As the legal framework between countries varies (e.g. EU GDPR regulation, article 35 and 36 about data protection impact assessment), important considerations need to be weighed and being able to express such constraints in machine readable form constitutes an important FAIRification task as it has a significant impact on data reuse possibilities.
FAIR implementation: Accessibility, open communication protocol standard(s) to control access.
FAIR implementation: Reusability, consider licences and consent to allow the re-use of data and metadata from a legal perspective. The FAIR cookbook recipe on representing permitted use using open standards (Declaring data permitted uses) provides a good starting point with hands-on examples.
Study conduct and data collection
Once a study is conducted and data collection initiated, more technical aspects come to the fore. How is the data referenced? How/where is data actually stored? Depending on the type of processes generating the data, different infrastructures might be used (see also “A multiplicity of clinical data types” in the Introduction of this guide). Primary resources for healthcare and clinical research can be identified as follows:
- Data generated as part of routine healthcare processes: Electronic Healthcare Records, Radiology, Pathology, Genomics, health insurance claims etc.
- Data generated as part of (interventional) clinical research: Clinical Trials / Studies
- Data generated for research and/or quality monitoring purposes: Observational Registries / Databanks / Biobanks
- Data generated by patients as part of self-monitoring via medical devices or patient reported outcomes.
As study sponsors often rely on contract research organisations (CRO) to perform the tasks of data collection, especially in the context of multicentric and multimodality studies, careful considerations should be made to properly specify how data should be provided and taken care of. This often means going down to the specifics of data format, controlled vocabulary choices and validation pipelines to ensure consistency from the start and avoid added downstream curation costs to reconcile different data sources. The topic of interacting with CRO is covered in a recipe available from the FAIR cookbook (Practical Considerations for CROs to play FAIR), which was contributed by Novartis AG.
FAIR implementation: Findability, an identifier strategy is essential to support future findability of data and metadata in a system (see Pistoia’s FAIRToolkit Adoption and Impact of an Identifier Policy as an example for an identifier strategy).
FAIR implementation: Interoperability, Convergence on terminology (e.g. LOINC for laboratory test coding).
Data curation / harmonisation processes
In a highly restricted and regulated environment, reviewing the data quality and curating the (raw) data is inevitable - even though data quality per se is not a dimension of FAIR (see publication “Maximizing data value for biopharma through FAIR and quality implementation: FAIR plus Q”). How much effort is needed to curate the associated metadata depends on the care and data entry validation procedures laid out during planning phases and while conducting the data collection of the study. Disparate clinical data sources might have to be harmonised for the purpose of healthcare quality assessments. Besides semantic interoperability, technical interoperability might be an obstacle when integrating multiple healthcare systems, across various geographical entities and jurisdiction in the case of multi-centric, multi-country trials. In such a setup, in order to comply with local regulations, data may have to be withheld.
FAIR benefit: Prospective FAIR data reduced the manual effort needed to curate datasets by embedding annotation requirements and validation rules in data acquisition systems and procedures have been shared and explained with involved parties, including CROs. (See Pistoia Alliance’s FAIR Toolkit use case, FAIRification of clinical trial data as an example for retrospective FAIRification. Also, see another use case Prospective FAIRification of Data on the EDISON platform as an example of prospective FAIRification of data).
Analysis of data
Analysis of the collected data might include the efficacy of drug and adverse events during the study or health and safety and signal detection. Complex analysis might involve multiple data sources or systems.
FAIR benefit: FAIR data significantly reduced the effort to integrate and combine data from different sources
Reporting
Clinical studies, if promising, are submitted for publication or to the authorities. After the primary submission, flexible reporting still is crucial. Companies have to engage with the authorities if there is e.g. a label change once the drug is on the market or when repurposing a drug.
FAIR benefit: Integrated data supports reporting and question answering
Data follow-up activities
The main purpose of the FAIR guiding principles is to enable and facilitate reusability of data. Follow up activities like sharing of data in private or public data catalogues, anonymisation of data or re-use of datasets for research purposes are very common use cases, which should be considered by stakeholders and in the data management plan.
FAIR benefit: Following the FAIR principles unlocks longevity of data and enables secondary use by providing clear data licences
Common Data Models for clinical data
In the previous chapter, we described a potential journey that creates a clinical dataset. The common data models play a huge role in storing and also exchanging data (see “Study conduct and data collection” in the previous chapter), thus we want to briefly dive into the most important standardisation systems that were already mentioned in the introduction:
- CDISC, which is essential for submitting clinical trials to regulatory authorities
- OMOP as sophisticated approach to store observational data
- FHIR, which focuses more on data exchange.
CDISC
The Clinical Data Interchange Standards Consortium (CDISC) is a standard developing organisation (SDO) which started as a volunteer group in 1997 with the goal of improving medical research and related areas of healthcare by working on the interoperability of data, resulting in a number of standards. Some of the CDISC standards are mandatory for submission to clinical regulators. Furthermore, CDISC is also cooperating with Health Seven International, abbreviated to HL7® . However, the long and rich history of CDISC may explain its complex landscape.
CDISC Operational Data Model (ODM)
The CDISC Operational Data model (ODM) is a standard designed for the exchange and archiving of translational research data and its associated metadata as well as administrative data, reference data and audit information. The ODM standard is used these days in many more ways than originally intended. Usage varies from Electronic data capture to planning, data collection and data analysis and archival. The implementation of the operational model is XML based (ODM-XML), which also facilitates the data exchange between different foundational standards as I was extended after its invention in 1999 for different purposes. ODM-XML is vendor-neutral and platform-independent format.
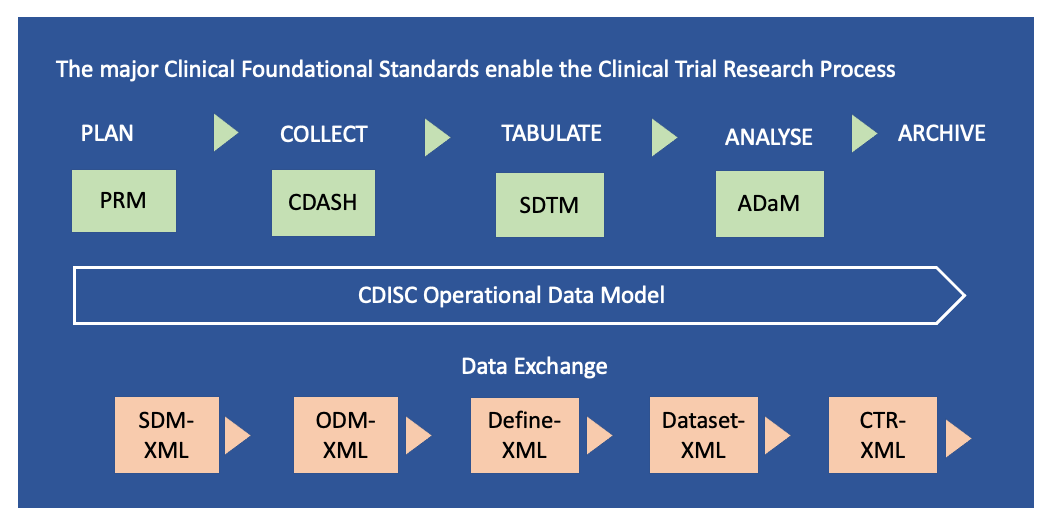 Figure 5: The CDISC clinical foundational standards consist of 1) Model for Planning (PRM), 2) Model for Data Collection (CDASH), 3) Model for Tabulation of Study Data (SDTM) and 4) Analysis Data Model (ADaM). These foundational standards are components of the CDISC Operational Data Model (ODM). The data exchange is facilitated through five extended markup languages (XML) which are 1) Study Trial Design (SDM), 2) Operational Data Model (ODM), 3) Dataset Metadata (Define), 4) Dataset Data (Dataset) and 5) Clinical Trial Registry (CTR). Adapted from: https://www.cdisc.org/standards and Current applications and future directions for the CDISC Operational Data Model standard: A methodological review
Figure 5: The CDISC clinical foundational standards consist of 1) Model for Planning (PRM), 2) Model for Data Collection (CDASH), 3) Model for Tabulation of Study Data (SDTM) and 4) Analysis Data Model (ADaM). These foundational standards are components of the CDISC Operational Data Model (ODM). The data exchange is facilitated through five extended markup languages (XML) which are 1) Study Trial Design (SDM), 2) Operational Data Model (ODM), 3) Dataset Metadata (Define), 4) Dataset Data (Dataset) and 5) Clinical Trial Registry (CTR). Adapted from: https://www.cdisc.org/standards and Current applications and future directions for the CDISC Operational Data Model standard: A methodological review
CDISC Foundational standards
The major “Foundational standards” are the building blocks of CDISC, as illustrated in Figure 5. They consist of protocols for the clinical research processes of planning (PRM - Protocol Representation Model), collecting data (CDASH), organising study data (SDTM) and analysis of data models (ADaM). In addition, there is a standard for tabulation of animal (non or pre-clinical) studies (SEND). In the following paragraphs we briefly describe the foundational standards, before we discuss the CDISC’s data exchange mechanism and the other important CDISC building blocks – the controlled terminology and so-called therapeutic area-specific extensions.
PRM
The Protocol Representation Model (PRM) is a way to “translate” the human readable research protocol into machine readable representation. Thus it focuses on study characteristics such as study design or eligibility criteria and notably also information that is necessary for submission to Clinical Trial Registries. PRM is aligned with the Biomedical Research Integrated Domain Group (BRIDG) Model (see section Shared model of clinical study design and outcome).
CDASH
Clinical Data Acquisition Standards Harmonisation (CDASH) is focused on the collection of data across studies with a focus on traceability between data collection and data submission to the regulatory agencies. Using CDASH during clinical trial data collection supports the creation of SDTM data for submission later on in the clinical data process. Or in other words, while the use of CDASH is not mandatory, it makes sense to stick to CDISC defined data elements from the start because it allows easier conversion to SDTM.
SDTM
The Study Data Tabulation Model (SDTM) is a standard method for structuring data about collecting, managing, analysing and reporting clinical study results. The format is table based, where each row represents an observation for a uniquely identifiable clinical study subject, e.g. by patientIdentifier, date, time, etc. Besides this identifying information, the row also contains one data point associated with the observation. This data format leads to large files with potentially many blank columns, though it does remain internally consistent. The headers in SDTM are standardised as they have fixed (variable) names and overlap to a certain extent with CDASH.
Different “domains” are represented in SDTM, for example the demographics domain (DM) or the subject visits domain (SV). Usually the data for each domain are stored in different files - the sum of these files represents the whole dataset of the clinical study.
ADaM
The Analysis data model (ADaM) is a subset of derived data from a SDTM dataset for statistical and scientific analysis. For example, the age of the patient is a derived variable and is calculated from the Date of Birth. Some ADaM variables are also specific/relevant to certain Indications (e.g. such as Progression Free Survival (PFS), Time To Progression (TTP)). The principles and standards specified in ADaM provide a clear lineage from data collection to data analyses.
Data Exchange
CDISC also provides standards for data exchange formats to enable the clinical research process, namely ODM-XML, which connects PRM, CDASH and SDTM. Define-XML and Dataset XML are ODM-XML extensions to connect the foundational standard SDTM to ADaM. Define-XML is also used to support the submission of clinical trials data to regulatory agencies, using a tabular dataset structure. Dataset XML complements Define XML by supporting the exchange of datasets based on Define-XML metadata.
CDISC controlled vocabulary and extensions
Controlled Terminology is the set of codelists and valid values used with data items within CDISC-defined datasets. Controlled Terminology provides the values required for submission to FDA and PMDA in CDISC-compliant datasets. Controlled Terminology does not tell you WHAT to collect; it tells you IF you collected a particular data item, how you should submit it in your electronic dataset. For example, if various lab tests were performed (e.g. WBC, glucose, albumin) CDISC provides vocabularies on how these tests should be reported, including the units of the measurements.
CDISC, in collaboration with the National Cancer Institute’s Enterprise Vocabulary Services (EVS), supports the Controlled Terminology needs of CDISC Foundational and Therapeutic Area Standards.
CDISC and FAIR
We can look at this at two levels. The first one is the FAIRness of the CDISC standards and specifications themselves. The second one is the FAIRness of CDISC coded data, i.e. clinical trial information recorded through an implementation of the CDISC standards stack.
FAIRness of CDISC standards
With the release of the CDISC Library, authorised users and vetted member organisations can get access the various CDISC specifications and their versions via a REST-API, the documentation of which is available here.
 Figure 6: The CDISC Library API Open API Specification compliant documentation. Note that accessing the full documentation requires an API key from the CDISC organisation.
Figure 6: The CDISC Library API Open API Specification compliant documentation. Note that accessing the full documentation requires an API key from the CDISC organisation.
The most interesting feature of the service is that it complies with the Open-API v3 specifications, which means that it allows software agents to rely and standardised annotations and API calls are self describing identifying parameters and response types.
The CDISC service thus ranks fairly highly in terms of FAIR maturity since it implements community standards for describing API. Responses from the CDISC Library REST endpoint are either json, xml, csv or excel format.
 Figure 7: Overview of the CDISC Library which can be found at https://www.cdisc.org/cdisc-library
Figure 7: Overview of the CDISC Library which can be found at https://www.cdisc.org/cdisc-library
FAIRness of CDISC coded clinical trial data
The different CDISC standards and exchange data formats are mostly table-based and are defining a grammar for structuring information in tables. They lack explicit semantics, and thus are not anchored to overarching ontologies/semantic models. This, in turns, may limit interoperability of CDISC coded clinical trial data with other data or clinical trial data coded using a different system, and thus FAIR maturity. To address this issue, a project called CDISC 360 was started. It tries to bring linked data and semantics to the CDISC standards. While this is an ambitious and promising effort, this is still very much work in progress.
Some of the challenges with these datasets include updating internal systems and providing guidance when SDTM versions change. Although there exists some standards (such as CDASH) for capturing the data, the standards do not cover all data types and extensions are made by the sponsors within the CDISC framework. Furthermore, capturing metadata about sponsor extensions is another challenge in itself.
Further reading on CDISC
- CDISC Collection at FAIRsharing
- CDISC Controlled Terminology and Glossary
- ODM and ODM-XML and ODM Implementation, CDISC data exchange
- Current applications and future directions for the CDISC Operational Data Model standard: A methodological review by Sam Hulme et al (2016) J Biomed Inf 60:352-362
- An assessment of the suitability of the Clinical Data Interchange Standards Consortium (CDISC) Operational Data Model (ODM) - J Biomed Inform. 2015 Oct; 57: 88–99.
OHDSI
Observational Health Data Sciences and Informatics (OHDSI, pronounced “Odyssey”) is an open-science community. The focus of OHDSI is on observational data, including real-world data (RWD) and real world evidence (RWE) generation. Its goal is improving health by empowering the community to collaboratively generate evidence that promotes better health decisions and better care. To enable this, the community established itself as a standard developing organisation (SDO) and started building information exchange specifications.
OMOP Common Data Model (OMOP CDM)
Under the OHDSI umbrella, the Observational Medical Outcomes Partnership (OMOP) common data model (CDM) was developed. The OMOP CDM is person-centric, as observational data capture what happens to a patient while receiving healthcare. Thus, all events are linked directly to the patient to allow a longitudinal view of the given healthcare and its outcome. This patient history, especially when summarised for a part of the population, can later be exploited for insights and for delivering improved health decisions and better care as stated in OHDSI’s mission statement.
Transforming data from different healthcare systems to converge to the OMOP CDM enhances interoperability and enables data analysis on combined data (meta-analysis).
The OMOP CDM uses standardised tables as its building blocks. There are tables defined for different areas, e.g. clinical data, vocabularies, metadata and health system tables. An overview of these tables can be found here and more details can be found here.
Besides the structure of the data in the described tables, OMOP CDM also uses standardised vocabularies to align the actual content - the meaning of the data (see next chapter). However, as noted with CDISC standards before, this suffers from the same issue of implicit semantics, where the meaning of relation between fields is unclear to software agents.
OHDSI standardised vocabularies
The common data model would not be able to fulfil its promise to boost interoperability without using agreed, standardised vocabularies underpinning it. However, even for controlled vocabularies, competing or conflicting standards exist, particularly within health care, where national and regional differences in classifications and healthcare systems (e.g. ICD10CM, UCD10GM), often mandated by regulatory agencies or e.g. insurance systems influence technical choices. OHDSI also improves the interoperability by mandating the use of standardised vocabularies within the CDM. These vocabularies include well known terminologies like International Statistical Classification of Diseases and Related Health Problems (ICD), SNOMED, RXNorm and LOINC (this is common to CDISC SDTM and offers a bridge to this clinical trial data stack of standards).
OHDSI defines a standardised vocabulary to try to bridge this gap. It is such a fundamental part of the system that the use of the standardised vocabulary is mandatory for every OMOP CDM instance. While OHDSI uses existing vocabularies, a common format is defined and third party vocabularies have to be transformed from their original format into the OHDSI vocabulary tables, most notably the concept table. A term from the selected vocabularies and ontologies is seen as an OMOP concept, stored and referred to by an OMOP concept id rather than its original identifier or URI. The original code (i.e its identifier) is also stored as the ‘concept_code’. Vocabulary concepts are arranged in domains and categorised as standard and non-standard, but the convention is to only use the standard concepts to refer to events. The relationships and hierarchical structure of the ontologies are stored in the OMOP vocabulary tables and can be used when analysing the data. The Athena website provides an environment to search and find entities in the hosted vocabularies.
Data analysis and evidence quality
After transforming data to the OMOP CDM, one can analyse the data using a variety of tools developed by the OHDSI community as open source software. The availability of a supporting suite of tools is one of the big advantages of this data model. It also includes tools for validating the quality of data.
Using the analytics tools, a range of use cases can be addressed, as indicated in the OHDSI book chapter “Data Analytics use cases”. After analysing data, the question arises how reliable is this evidence? This is also addressed in detail in the OHDSI book in the chapter “Evidence quality”.
OHDSI study
A study is represented in OHDSI by a study protocol and a so-called study package. The protocol is a human readable description of the observational study and should contain all necessary information to be able to reproduce the study by, e.g. including details about the study population or the methods or statistical procedures used throughout the study. The study package is a machine-readable implementation of the study. OHDSI supports this through tools for planning, documenting and reporting for observational studies. The data needs to be transferred/translated into the OMOP common data model, which we described previously. Despite all these measures, the reproducibility is not guaranteed and this is a point of discussion (this video gives more information on this problem). For further reading on OHDSI studies, we refer to this chapter in The Book of OHDSI.
OHDSI standards and FAIR
As done when assessing CDISC standards, let’s look at both the FAIRness of the specifications themselves and that of the datasets.
FAIRness of OHDSI standards
All OHDSI development is available openly from a GitHub organisation, with dedicated repositories for the standard specifications, the vocabulary or the supporting tools (e.g. https://github.com/OHDSI/DataQualityDashboard). Each of these repositories specify a licence (e.g. Apache 2.0 for CDM or the Data Quality Dashboard and the ‘unlicense licence’ for the vocabulary), which is key to establish Reuse. In spite of being hosted on GitHub, it seems that releases have not been submitted to CERN’s Zenodo document archive to obtain digital object identifier. Doing so would increase findability as it would create a record with associated metadata and version information. OHDSI standards have records in the FAIRsharing.org catalogue (see OHDSI Vocabulary and here for OMOP CDM).
FAIRness of OHDSI standards encoded observational data
Observational data coded using the OHDSI standards stack provides a high potential for reuse through interoperability delivered by the use of a common syntax (OMOP CDM) and vocabularies (OMOP Vocabulary and ATHENA tools). However, as seen before in the clinical context with the CDISC standards, a number of features hamper full machine actionability and FAIRness. First, it is the lack of a semantic model representing the relations between OMOP CDM entities and fields. Then, due to sensitivity of the data stored in that format, accessing data is under the control of the data access committee. Making patient centric information findable requires careful considering, from data protection impact assessment to proper managed access brokering. However, this is beyond the scope of this guide so no further details will be given.
It could be that a GA4GH beacon (link) like approach could be considered for interrogating repositories of OHDSI coded datasets. Beacon is an API (sometimes extended with a user interface) that allows for data discovery of genomic and phenoclinic data.
Bearing this in mind, it is worth noting that the OHDSI group provides several “test” datasets, ranging from synthetic data to public, anonymised data for the purpose of training users and developers with the standards and the practice. These are important resources as they can be used to assess FAIR maturity using untethered data and perform theoretical work which could benefit the entire community.
The OMOP CDM was developed originally for US insurance claims data and was later adapted for hospital records, in general by mapping other data sources (e.g. EU data from general practitioners) to the OMOP CDM, which can be a resource intensive task. For instance, mapping of local source codes to the standard vocabularies used within the OHDSI community is time intensive and can lead to loss of nuance and details that are in the source data. For semantic mappings, the source data are stored in the database, however those values can’t be used in the analysis tools. On the other hand, extensions for the model have been developed to accommodate oncology data (episode tables in OMOP CDM v5.4) and are being developed for clinical trial data.
Further reading on OHDSI
- OMOP record in FAIRsharing and OHDSI vocabularies record in FAIRsharing
- Book of OHDSI
- Standardised vocabularies used by OMOP CDM. Available through Athena. Updates on Github. OHDSI standardised vocabularies (link)[https://www.ohdsi.org/web/wiki/doku.php?id=documentation:vocabulary:sidebar]
FHIR®
FHIR® (Fast Healthcare Interoperability Resources) is a specification for health care data exchange, published by HL7® which is Health Seven International which is a not-for-profit, ANSI-accredited standards developing organisation, like CDISC Foundation. HL7 provides a comprehensive framework and related standards for the exchange, integration, sharing and retrieval of electronic health information that supports clinical practice and the management, delivery and evaluation of health services.
FHIR is an interoperability standard intended to facilitate the exchange of healthcare information between healthcare providers, patients, caregivers, payers, researchers, and anyone else involved in the healthcare ecosystem. It consists of 2 main parts – a content model in the form of ‘resources’, and a specification for the exchange of these resources (e.g messaging and documents) in the form of real-time RESTful interfaces. Thus, technically FHIR is implemented as a (RESTful) API service, which allows exposing discrete data elements instead of documents and therefore allows granular access to information and flexibility in data representation formats (e.g. JSON, XML or RDF).
The home page for the FHIR standard presents the five levels of the standard/framework. FHIR defines the “basics” at level 1, “basic data types and metadata”, to represent information. Other levels include “implementation and binding to external specifications” on level 2, or “linking to real world concepts in the healthcare system” on level 3. Within each level, different FHIR modules are defined, e.g. clinical or diagnostics but also workflows or financials, so the FHIR specification covers all sorts of (clinical) information and its exchange. The building blocks of FHIR are “FHIR resources” which can be extended if needed, and exchanged through standardised mechanisms as mentioned above
FHIR and FAIR
- The initial release of the FHIR for FAIR Implementation Guide aims to provide guidance on how HL7 FHIR can be used for supporting FAIR health data at the study level, for similar reasons to this guide. We refer to the FHIR for FAIR Implementation Guide for further details: FHIR for FAIR Home Page
In the context of real world evidence (RWE), the FHIR standards are a powerful enabler for interoperability. Furthermore, both OHDSI and CDISC now offer mappings for alignment to elements of FHIR.
It is noteworthy that FHIR and CDISC recommend or mandate similar common semantic resources (e.g. RxNorm, LOINC for drug treatments and laboratory tests description), so that semantic and syntactic convergence are beginning to emerge.
Further reading on FHIR
Shared model of clinical study design and outcome
In the sections above, we explored different common ways to store and exchange clinical trial and RWD data, namely CDISC, OHDSI and FHIR respectively. They all serve a different purpose and come with different advantages and limitations. However, each model does contain similar and often identical concepts, expressed, stored and handled in different ways. If one takes a step back, there is a shared semantic model, which could greatly help align data sets coming from different sources. As interoperability is a major aspect of FAIR, so is alignment between different approaches.
The BRIDG initiative
One such effort is the BRIDG initiative, see figure 8 for the high level concept map that is used to convert to a common semantic model. For the scope of this guide, concepts related to the study are most relevant, e.g. study site, organisation, protocol, eligibility criteria, arm.
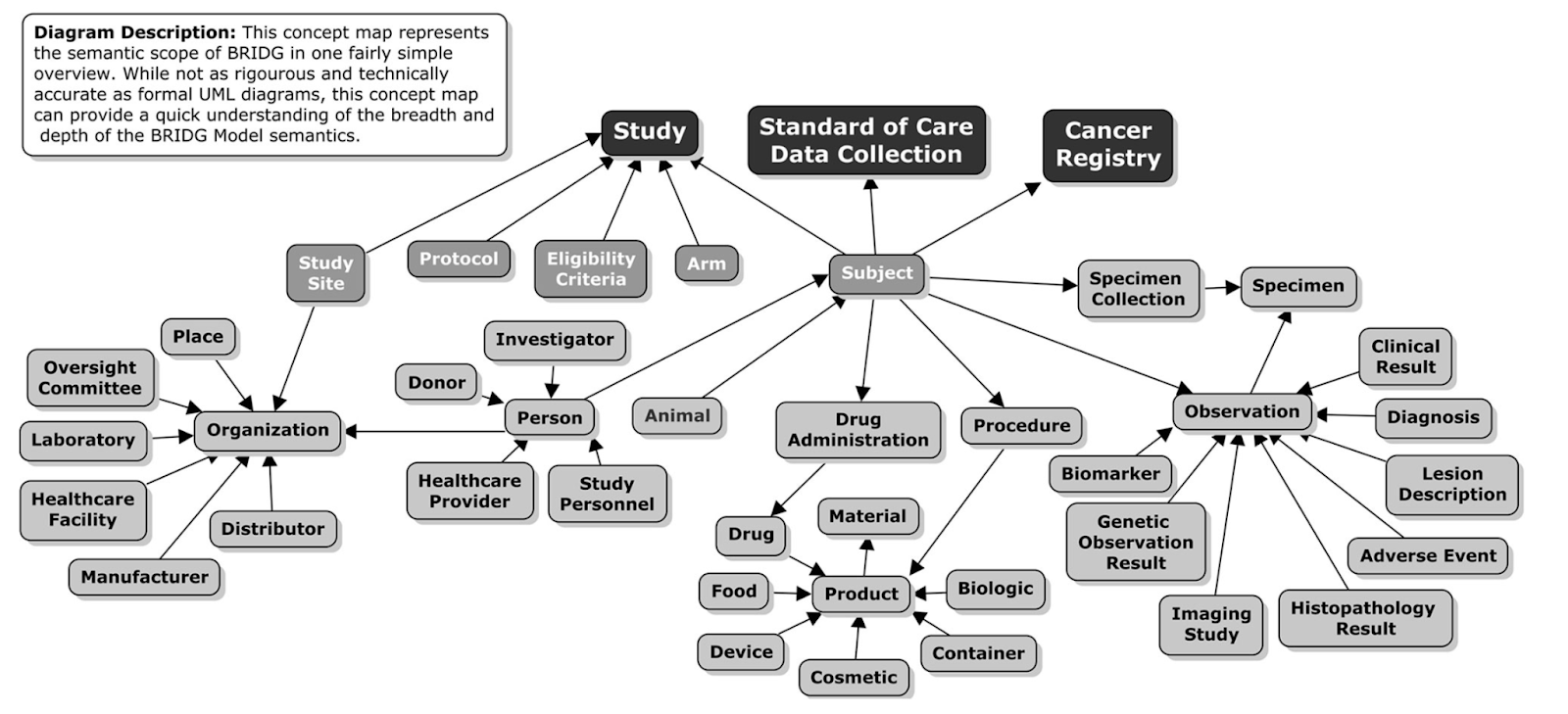 Figure 8: Taken from https://bridgmodel.nci.nih.gov/high-level-concept - showing the BRIDG High Level Concept Map.
Figure 8: Taken from https://bridgmodel.nci.nih.gov/high-level-concept - showing the BRIDG High Level Concept Map.
BRIDG stands for Biomedical Research Integrated Domain Group and tries to map different domain data models (aka common data models or CDMs) to higher level concepts and align those via the BRIDG model. The BRIDG Domain Information Model can be searched and studied in great detail, e.g. see figure 8 for the BRIDG view for “Structured Protocol - Backbone”.
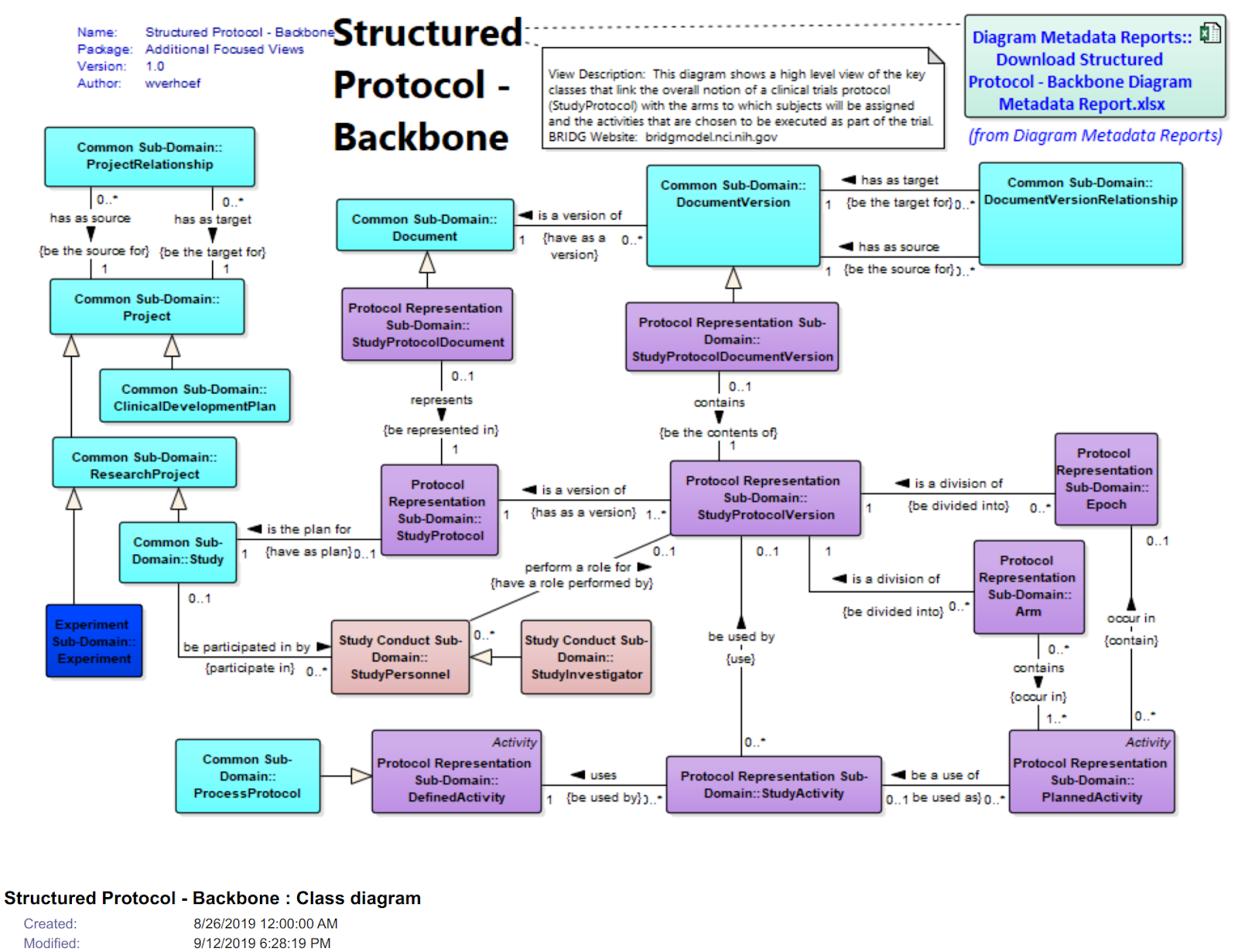 Figure 9: View of the “Structured Protocol - Backbone” in the BRIDG Browser
Figure 9: View of the “Structured Protocol - Backbone” in the BRIDG Browser
Table 3: Semantic mappings using NCIT to CDISC, OMOP and FHIR. Please note that OMOP tables are very patient centric and study elements are not modelled per se in the CDM.
| Semantic Concept | NCIT definition | NCIT URI | CDISC domain & data element | OMOP table & data element | FHIR resource & data element |
|---|---|---|---|---|---|
| Study Identifier | A sequence of characters used to identify, name, or characterise the study. | NCIT_C83082 | ODM/Study OID | ? | ResearchStudyidentifier |
| Study Name | The name applied to a scientific investigation | NCIT_C68631 | ODM/StudyName | ? | ResearchStudyname/title |
| Study Start | The official beginning of a clinical study, as specified in the clinical study report | NCIT_C142714 | SDM/StudyStart | ? | ResearchStudyperiod |
| Study Site Identifier | A sequence of characters used to identify, name, or characterize the study site | NCIT_C83081 | DM/SITEID | care sitecare_site_id | OrganizationIdentifier |
| Ethnic Group | A social group characterized by a distinctive social and cultural tradition that is maintained from generation to generation… | NCIT_C16564 | DM/ETHNIC | Personethnicity_concept_id | Patientextension: us-core-ethnicity |
| Sex | The assemblage of physical properties or qualities by which male is distinguished from female; the physical difference between male and female; the distinguishing peculiarity of male or female. | NCIT_C28421 | DM/Sex | Persongender_concept_id | Patientgender |
Normalisation and overlaps FHIR/OHDSI/CDISC
Besides the discussed BRDIG efforts described above, there are a few other initiatives going on trying to map between OHDSI, FHIR and CDISC which we shall mention here briefly. The IMI EHDEN project looks at the OMOP interoperability to different systems (See EHDEN roadmap). Working groups were established, e.g ODHSI/FHIR (link, OMOP2FHIR mappings gsheet) or a OMOP/CDSIC working group (link).
Also HL7 FHIR looks into mapping to other approaches (link), besides OMOP e.g. to PCORNet, i2b2/ACT. See the detailed specifications of the mappings here.
CDISC released the “FHIR to CDISC Joint Mapping Implementation Guide v1.0”, which can be found here.
Semantic integration with Semantic Web standards
As more and more systems serve data through application programmatic interface delivering JSON payloads, this opens the possibility of semantic integration by mapping the entities and objects served to a common semantic framework.
Projects such as FHIR JSON-LD playground or CDISC Phuse RDF offer the possibility of building JSON-LD context file which would provide the semantic context needed to turn JSON message into a JSON-LD one. While the technology is available and mature, the availability of an overarching semantic model/ontology that would serve the same purpose as the BRIDG model did in the UML world remains to be established.
